



Retinitis pigmentosa (RP) ist eine Gruppe seltener Augenkrankheiten, die die Netzhaut – die lichtempfindliche Gewebeschicht auf der Rückseite des Auges – betreffen. RP führt dazu, dass die Zellen der Netzhaut im Laufe der Zeit langsam absterben, was zu einem Sehverlust führt.
RP ist eine genetisch bedingte Krankheit, mit der Menschen geboren werden. Die an der RP beteiligten Genmutationen können von einem oder beiden Elternteilen an ein Kind weitergegeben werden. Manchmal tritt RP jedoch auch sporadisch auf, d. h. es gibt keine erkennbare Familienanamnese.
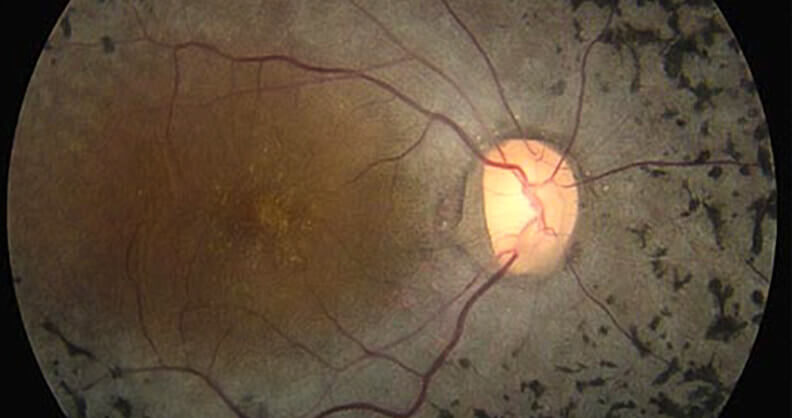
Die Symptome treten in der Regel in der Kindheit auf, und die meisten Menschen verlieren schließlich den größten Teil ihrer Sehkraft. Es gibt keine Heilung für RP. Aber Sehhilfen und Rehabilitationsprogramme (Training) können Menschen mit RP helfen, das Beste aus ihrer Sehkraft zu machen.
Sie sollten jedoch wissen, dass Retinitis pigmentosa nur eine Form der Retinitis ist, und dass Sie auch eine infektiöse oder andere Art von Retinitis bekommen können.
Was ist Retinitis Pigmentosa?
In den meisten Fällen wird RP durch Veränderungen in den Genen verursacht, die die Zellen in der Netzhaut steuern. Diese veränderten Gene werden von den Eltern an die Kinder weitergegeben.
Die Netzhaut enthält Zellen, die so genannten Fotorezeptoren, die Licht absorbieren und in elektrische Signale umwandeln. Diese Signale werden dann über den Sehnerv an das Gehirn weitergeleitet, damit dieses sie zu den Bildern verarbeiten kann, die der Mensch sieht.
Die Fotorezeptoren bestehen aus Stäbchen und Zapfen. Die Stäbchen sind für das Sehen bei schlechten Lichtverhältnissen verantwortlich. Die Zapfen helfen dem Menschen, Farben und Details zu erkennen.
Zunächst beeinträchtigt die RP die Stäbchen, was zu Problemen beim Nachtsehen führt. Wenn die Stäbchen stärker geschädigt werden und absterben, wirkt sich die RP auch auf die Zapfen aus und beeinträchtigt die Sehkraft weiter. Weltweit ist 1 von 4.000 Menschen von RP betroffen.
Retinitis-Pigmentosa-Symptome und -Verlauf
RP ist eine fortschreitende Erkrankung, d. h. sie verschlimmert sich im Allgemeinen mit der Zeit. Die Symptome treten typischerweise in der Kindheit auf. Das Erkrankungsalter ist sehr unterschiedlich und liegt zwischen dem 25. und 40. Lebensjahr.
Es gibt Fälle, in denen die Betroffenen unter 20 Jahre alt sind, und seltener Fälle, in denen die Symptome erst im Alter von über 50 Jahren auftreten.Mit fortschreitender RP können neue oder sich verschlechternde Symptome auftreten, die das Sehvermögen beeinträchtigen.
Symptome im Frühstadium
Im Frühstadium der Erkrankung sind die Stäbchen stärker betroffen als die Zapfen. Da die Stäbchen absterben, können die ersten Anzeichen und Symptome folgende sein
- Schwierigkeiten bei der Anpassung an veränderte Lichtverhältnisse
- Photophobie, d. h. Empfindlichkeit gegenüber hellem Licht
- Schwierigkeiten beim Sehen in einem abgedunkelten Raum.
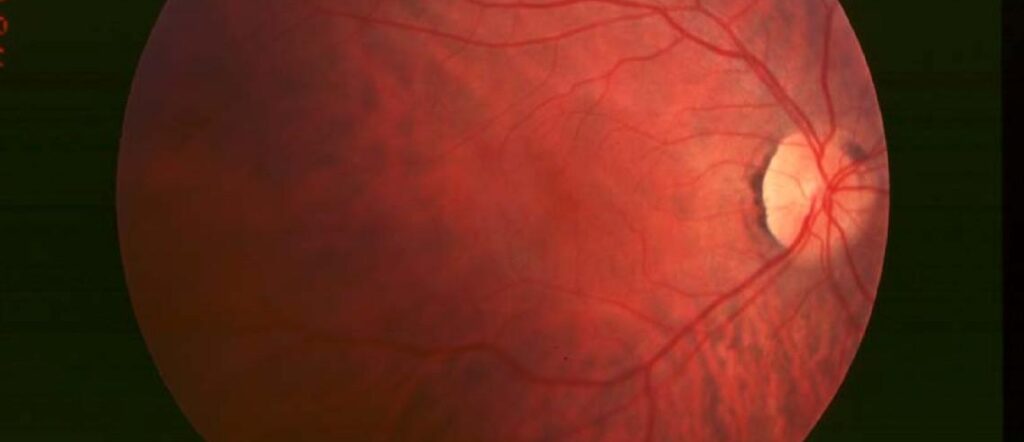
Symptome im Spätstadium
Wenn die Erkrankung fortschreitet und auch die Zapfen absterben, kommt es zu einem allmählichen Verlust des Gesichtsfelds und zu einem Tunnelblick. Schließlich verliert eine Person, die mit RP lebt, den größten Teil ihres Sehvermögens.
Der Verlauf der RP kann von Mensch zu Mensch sehr unterschiedlich sein. Manche Menschen verlieren ihr Sehvermögen schon in jungen Jahren, andere behalten ihr zentrales Sehvermögen bis weit in ihre 50er Jahre hinein.
Das zentrale Sehvermögen ist für das Sehen von Details verantwortlich und wird für Aufgaben wie Lesen oder Einfädeln einer Nadel benötigt.
Zu den Anzeichen und Symptomen einer Retinitis pigmentosa im fortgeschrittenen Stadium gehören im Allgemeinen:
- Das Gefühl von blinkendem oder blitzendem Licht.
- Tunnelblick (nur zentrales Sehen).
- Empfindlichkeit gegenüber hellem Licht oder Unbehagen bei hellem Licht (Photophobie).
- Verlust der Fähigkeit, Farben zu sehen.
- Sehr geringes Sehvermögen.
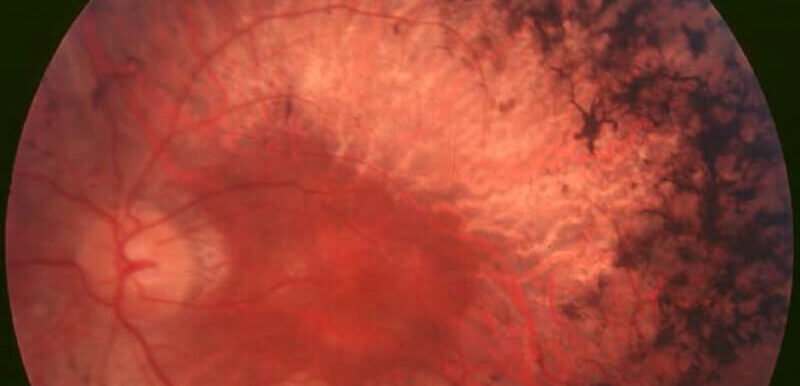
Retinitis Pigmentosa Wie Lange Bis Zur Erblindung
Während einige Patienten mit Retinitis pigmentosa bis zum Alter von 30 Jahren praktisch erblindet sind, behalten andere ihr Sehvermögen bis zum Alter von 80 Jahren oder darüber hinaus.
Oft sind beide Augen von einem ähnlichen Sehverlust betroffen. Die RP ist jedoch eine langsam fortschreitende Krankheit, die sich über viele Jahre hinzieht, und die meisten Patienten erblinden nie vollständig.
Die Retinitis pigmentosa beginnt stumm und langsam, so dass die Patienten normalerweise erst 15 Jahre nach Beginn der Nachtblindheit einen Augenarzt aufsuchen.
Nachtblindheit sollte also ein Alarmsignal für Sie sein, damit Sie Ihren Augenarzt aufsuchen.
Retinitis Pigmentosa Vererbung
RP ist eine genetisch bedingte Erkrankung. Sie kann aufgrund einer Mutation in über 50 verschiedenen Genen auftreten, die für die Anweisungen zur Herstellung von Proteinen verantwortlich sind, die in den Photorezeptoren benötigt werden. Eltern können RP auf drei Arten an ihre Nachkommen weitergeben:
Autosomal-rezessive Vererbung
Wenn zwei Menschen, die bestimmte rezessive Genmutationen tragen, ein Kind bekommen, besteht eine:
- 1 zu 4 Chance, dass das Kind RP entwickelt
- 1 zu 2 Chance, dass das Kind Träger von RP wird
- 1 zu 4 Chance, dass das Kind keine RP entwickelt oder Träger von RP wird
Autosomal-dominante Vererbung
Wenn ein biologischer Elternteil die Genmutation auf der dominanten Version eines Gens trägt, braucht das Kind nur eine Kopie der Mutation, um RP zu entwickeln. Die Wahrscheinlichkeit, an RP zu erkranken, liegt dann bei 1:2.
X-chromosomale Vererbung
Dies bedeutet, dass sich das mutierte Gen auf dem X-Chromosom befindet. Männer haben in der Regel ein X-Chromosom, während Frauen zwei haben. Frauen erkranken nicht an der X-chromosomal vererbten RP, da das zweite X-Chromosom die Wirkung aufhebt.
Wenn ein weiblicher leiblicher Elternteil Träger der X-chromosomalen RP ist, hat ein männliches Kind eine Chance von 1 zu 2, an RP zu erkranken, und ein weibliches Kind hat eine Chance von 1 zu 2, Träger zu werden.
Studien deuten darauf hin, dass etwa 20 % der RP-Fälle autosomal rezessiv sind, 10-20 % autosomal dominant, 10 % X-chromosomal rezessiv und der Rest sporadisch
Diagnose von Retinitis Pigmentosa
Ein erfahrener Augenarzt kann im Rahmen einer umfassenden Untersuchung des erweiterten Auges nach RP suchen. Die Untersuchung ist einfach und schmerzlos.
Der Arzt gibt Ihnen einige Augentropfen, um Ihre Pupille zu erweitern, und untersucht dann Ihre Augen auf RP und andere Augenprobleme. Die Untersuchung umfasst auch einen Gesichtsfeldtest, um das periphere (seitliche) Sehen zu überprüfen.
Zu den Tests, mit denen sowohl die Diagnose als auch der Schweregrad der RP ermittelt werden kann, gehören:
- Elektroretinographie: Bei diesem Test wird die elektrische Aktivität des Auges gemessen, um festzustellen, wie gut die Netzhaut auf Licht reagiert.
- Genetische Tests: Der Betroffene gibt eine Gewebe- oder Blutprobe ab, um nach einem verantwortlichen Gen zu suchen und die Erfolgsaussichten einer Gentherapie zu beurteilen.
- Optische Kohärenztomographie: Dabei wird ein hochauflösendes Bild der Netzhaut aufgenommen, um die Diagnose zu stellen und festzustellen, wie sich die RP auf das Auge der Person auswirkt, insbesondere auf die Makula, die für das zentrale Sehen verantwortlich ist.
- Gesichtsfeldtest: Damit werden blinde Flecken im Sehvermögen einer Person gesucht und gemessen.
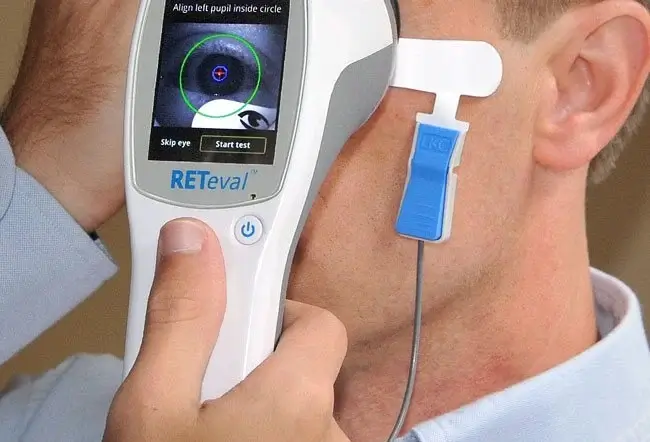
Retinitis Pigmentosa: Wann zum Arzt?
In der Regel sollten Sie Ihren Augenarzt regelmäßig aufsuchen. Wenden Sie sich immer an ihn, wenn Sie neue oder sich verschlimmernde Symptome haben, z. B:
- Ein zunehmender Verlust der Sehkraft, entweder in Bezug auf die Klarheit oder die Farbe.
- Ein neues Gefühl von Unbehagen oder Schmerzen.
Wie bereits beschrieben, suchen die Patienten ihren Augenarzt in der Regel erst auf, wenn viel Zeit vergangen ist. Stellen Sie sicher, dass Sie Ihren Arzt aufsuchen, wenn die ersten Anzeichen der RP auftreten, z. B. Nachtblindheit.
Retinitis-Pigmentosa-Behandlung (Therapie)
Derzeit gibt es keine Heilung oder eine einzige empfohlene Behandlungsmethode für RP. Ein Arzt kann die folgenden Maßnahmen empfehlen, um die Krankheit in den Griff zu bekommen:
- Augentropfen: Dorzolamid-Tropfen können die Schwellung des Makulaödems, das sich bei Retinitis pigmentosa entwickelt, reduzieren.
- Medikamente: Acetazolamid (Diamox), ein harntreibendes Medikament, kann ebenfalls bei Makulaödemen helfen.
- Chirurgie: Wenn eine Person mit RP Katarakte entwickelt, kann eine Kataraktoperation helfen, das Sehvermögen zu verbessern.
- Verwendung von Sehhilfen und Hilfsmitteln. Es gibt eine Reihe von Lupen und Technologien, die Dinge oder Personen erkennen können, auf die der Träger zeigt.
- Verwendung von Sonnenbrillen und anderen Methoden, um zu viel Licht zu vermeiden. Licht kann die RP verschlimmern.
Forscher arbeiten daran, mehr über die Gene herauszufinden, die RP verursachen. Ziel ist es, auf der Grundlage der Gene einer Person Maßnahmen zu entwickeln, die das Fortschreiten der Krankheit aufhalten oder umkehren können.
Einige Forschungsarbeiten haben gezeigt, dass Vitamin A das Fortschreiten bestimmter Formen von RP verlangsamen kann, aber es gibt Bedenken, dass eine hohe Einnahme dieser Nahrungsergänzungsmittel zu einer Verschlimmerung anderer Augenerkrankungen führen kann.
Ihr Augenarzt kann Sie über die Risiken und Vorteile von Vitamin A beraten und darüber, wie viel Sie unbedenklich einnehmen können. Die Einnahme von zu viel Vitamin A kann schädlich sein, und es gibt keine stichhaltigen Beweise für seine Wirkung auf das Fortschreiten der RP. Daher werden Vitamin-A-Präparate derzeit nicht empfohlen.
Außerdem sollten Menschen mit Sehschwäche einen Rehabilitationsprozess für ihr Sehvermögen einleiten. Spezialisierte Fachleute können dabei helfen, indem sie Dienstleistungen wie Ergotherapie anbieten. Die American Foundation for the Blind (Amerikanische Stiftung für Blinde) bietet ebenfalls verschiedene Ressourcen für Erwachsene, die zum ersten Mal mit einer Sehschwäche konfrontiert sind.

Kann ich Retinitis Pigmentosa vorbeugen?
Da die meisten Formen von Retinitis pigmentosa vererbt werden, können Sie RP nicht verhindern. Sie können jedoch Maßnahmen ergreifen, um Ihre Augen so gesund wie möglich zu erhalten:
- Regelmäßige Termine bei Ihrem Augenarzt vereinbaren und einhalten.
- eine Sonnenbrille tragen und helles Licht meiden.
- Sie sollten sich so gesund wie möglich ernähren und Sport treiben.
Ausblick
Menschen, die mit RP leben, werden mit der Zeit an Sehkraft verlieren. Die Geschwindigkeit des Fortschreitens und das Ausmaß des Sehverlusts können von Mensch zu Mensch unterschiedlich sein.
Die Aussichten hängen vom Muster der Vererbung ab. Zum Beispiel:
- Die autosomal rezessive Form der RP kann zu einem frühen Auftreten der Symptome führen und schwere Nachtblindheit und Sehkraftverlust verursachen.
- Die X-chromosomal rezessive RP ist mit dem schwersten Sehverlust verbunden.
- Die autosomal-dominante Form ist die am wenigsten schwere Form. Sie kann zu einem allmählichen Auftreten der Symptome führen, sobald die Betroffenen das Erwachsenenalter erreicht haben.
- Bei allen Formen kommt es zu einem Tunnelblick, und die meisten Menschen mit RP werden irgendwann erblindet sein. Ein völliger Verlust des Sehvermögens ist jedoch ungewöhnlich.
Menschen, die mit RP leben, sollten mit einem Arzt sprechen, um einen Plan für den Umgang mit ihrem Sehverlust zu entwickeln und möglicherweise das Fortschreiten der Krankheit zu verlangsamen.